CLASHChimeras¶
CLASHChimeras is a Python package for analysing CLASH datasets. It takes raw fastq files as input and provides comprehensive analysis of RNA profiles and chimeric reads identification. The output is CSV and BED format files for easy visualization in Genome Browsers.
Installation¶
You can install it using pip after you have setup Python version 3.4 or above. Please use this guide for setting up Python if you have not done it already. After setting up Python and pip, you can run this on your shell
For local installation (Usually $HOME/.local):
$ pip3 install --user CLASHChimeras
For global installation (Usually /usr/):
Note
You should have sudo privileges
$ sudo pip3 install CLASHChimeras
Dependencies¶
Warning
These dependencies must be satisfied if you want to use align-for-chimeras
CLASHChimeras requires certain software to be installed and setup before you can use it completely. The software you need to explicitly install are the following:
Usage¶
The package can be used by three executable scripts:
download-for-chimeras¶
align-for-chimeras¶
Warning
The input fastq is expected to be adapter trimmed and quality controlled
Note
Flexbar can be used to trim raw fastq sequences
find-chimeras¶
Note
It’s recommended that you provide SAM files as input which are generated using align-for-chimeras
Todo
Provide support for detecting chimeras between same RNA types
Example¶
We will be using the a dataset from CLASH experiment which is hosted here
In this instance, we’ll be using the first 4 million reads from the dataset. The sequential order to find chimeras on CLASH datasets using this package is the following:
Run download-for-chimeras¶
Run download-for-chimeras for the first time to download sequences and generate necessary indexes
The dataset that we are using here belong to H. sapiens. The sequence database needs to be downloaded from Gencode and miRBase. Here’s how you can download:
The code below assumes the default path as ~/db/CLASHChimeras but if you
want a different folder to put your sequences, please specify it using
--path /path/to/your/folder as a argument. It’s highly recommended to
get yourself familiar with the arguments by typing download-for-chimeras -h
$ download-for-chimeras -gor "H.sapiens" -mor hsa
Note
It’s an interactive script which prompts for user input when selecting the release version.
Warning
Please be patient as this is a big download and index generation takes even longer
Warning
The latest release from Gencode when downloaded and after all indexes are generated, takes around 11G of space
Below is an example of how download-for-chimeras runs.
Note
All the database files are already present in this example run, so they are verified by sha256sums. Thus, the timestamps are very close to each other. Actual download and generation of indexes will take a while
Indexes¶
There are a series of bowtie2 and tophat indexes generated after you’ve run
download-for-chimeras script. Assuming that you ran the command below and
selected the latest versions of Gencode and miRBase, the following indexes
will be generated automatically
$ download-for-chimeras -gor "H.sapiens" -mor hsa
smallRNA & targetRNA Indexes¶
These indexes can be used as --smallRNAIndex -si or --targetRNAIndex
-ti in align-for-chimeras
| Path for index | Index Type | RNA Type |
|---|---|---|
| ~/db/CLASHChimeras/Gencode/H.sapiens/22/gencode.v22.pc_transcripts | Bowtie2 | protein_coding |
| ~/db/CLASHChimeras/Gencode/H.sapiens/22/gencode.v22.lncRNA_transcripts | Bowtie2 | lncRNA |
| ~/db/CLASHChimeras/Gencode/H.sapiens/22/gencode.v22.snoRNA_transcripts | Bowtie2 | snoRNA |
| ~/db/CLASHChimeras/Gencode/H.sapiens/22/gencode.v22.snRNA_transcripts | Bowtie2 | snRNA |
| ~/db/CLASHChimeras/Gencode/H.sapiens/22/gencode.v22.tRNA_transcripts | Bowtie2 | tRNA |
| ~/db/CLASHChimeras/Gencode/H.sapiens/22/gencode.v22.misc_RNA_transcripts | Bowtie2 | misc_RNA |
| ~/db/CLASHChimeras/Mirbase/21/hsa-hairpin | Bowtie2 | miRNA-hairpin |
| ~/db/CLASHChimeras/Mirbase/21/hsa-mature | Bowtie2 | miRNA-mature |
Genome-Index¶
This index should be provided if you run align-for-chimeras with
--run tophat
| Path for index | Type |
|---|---|
| ~/db/CLASHChimeras/Gencode/H.sapiens/22/GRCh38.p2.genome | Bowtie2 |
Transcriptome-Index¶
This index should be provided if you run align-for-chimeras with
--run tophat along with Genome-Index
| Path for index | Type |
|---|---|
| ~/db/CLASHChimeras/Gencode/H.sapiens/22/gencode.v22.chr_patch_hapl_scaff.annotation | tophat |
Annotation¶
| Annotation File | RNA type |
|---|---|
| ~/db/CLASHChimeras/Gencode/H.sapiens/22/gencode.v22.chr_patch_hapl_scaff.annotation.gtf | protein_coding |
| ~/db/CLASHChimeras/Gencode/H.sapiens/22/gencode.v22.chr_patch_hapl_scaff.annotation.gtf | lncRNA |
| ~/db/CLASHChimeras/Gencode/H.sapiens/22/gencode.v22.chr_patch_hapl_scaff.annotation.gtf | snRNA |
| ~/db/CLASHChimeras/Gencode/H.sapiens/22/gencode.v22.chr_patch_hapl_scaff.annotation.gtf | snoRNA |
| ~/db/CLASHChimeras/Gencode/H.sapiens/22/gencode.v22.chr_patch_hapl_scaff.annotation.gtf | misc_RNA |
| ~/db/CLASHChimeras/Gencode/H.sapiens/22/gencode.v22.tRNAs.gtf | tRNA |
| ~/db/CLASHChimeras/Mirbase/21/hsa.gff3 | miRNA |
Run align-for-chimeras¶
Note
Please refer to Indexes when selecting --smallRNAIndex -si or
targetRNAIndex -ti when you run align-for-chimeras
For this instance, we want to find the chimeras between miRNA and
protein_coding from the raw reads. After you have successfully run
download-for-chimeras and made sure that all the indexes are present for
your alignment to begin, please use the following command
$ align-for-chimeras -i E3_4M.fastq.gz -gz -r bowtie2 -si ~/db/CLASHChimeras/Mirbase/21/hsa-hairpin -ti ~/db/CLASHChimeras/Gencode/H.sapiens/22/gencode.v22.pc_transcripts -o E3-miRNA-pc
This is how it runs.
After the successful execution of align-for-chimeras, these are the files
that are generated
- E3-miRNA-pc.smallRNA.sam
- E3-miRNA-pc.targetRNA.sam
Note
Please use --threads -n to specify the number of cores to use
when executing Bowtie2
align-for-chimeras also provides an argument to run tophat as well. This
helps in visualise the transcript coverage across the genome. Please use the
following command to align to the whole genome
$ align-for-chimeras -i E3_4M.fastq.gz -gz -r tophat -gi ~/db/CLASHChimeras/Gencode/H.sapiens/22/GRCh38.p2.genome -tri ~/db/CLASHChimeras/Gencode/H.sapiens/22/gencode.v22.chr_patch_hapl_scaff.annotation -o E3_4M
To create bigWig file from the tophat output, I’d recommend using deepTools to create normalized coverage file from the following wiki page
Let’s move forward with finding chimeras between these RNA types
Run find-chimeras¶
Note
Please refer to Annotation when selection --smallRNAAnnotation
-si or --targetRNAIndex -ti when you run find-chimeras
Following up after running align-for-chimeras, it’s time to detect chimeras.
Please make sure that you have the SAM files generated from
align-for-chimeras, please use the following command
$ find-chimeras -s E3-miRNA-pc.smallRNA.sam -t E3-miRNA-pc.targetRNA.sam -ggs -sa ~/db/CLASHChimeras/Mirbase/21/hsa.gff3 -ggt -ta ~/db/CLASHChimeras/Gencode/H.sapiens/22/gencode.v22.chr_patch_hapl_scaff.annotation.gtf -o E3-miRNA-pc
This is how the above command runs
After the successful execution of find-chimeras, these are the files that
are generated
- E3-miRNA-pc.chimeras.tsv
- E3-miRNA-pc.smallRNA.bed
- E3-miRNA-pc.targetRNA.bed
Note
Please note if you have not specified
--getGenomicLocationsSmallRNA -ggs, <sample>.smallRNA.bed will not be
generated. If you haven’t specified --getGenomicLocationsTargetRNA -ggt,
<sample>.targetRNA.bed will not be generated.
You can view the chimeras from the <sample>.chimeras.tsv file that is
generated. If you want to visualize the data in genome browsers, you can do
that by adding the <sample>.smallRNA.bed and <sample>.targetRNA.bed
in the IGV or your genome browser of choice.
Note
Please check the genome assembly version described in Genome-Index and make sure you have the same or corresponding version set in your genome browser
Possible combinations¶
Because of the modular design of the software, it is possible to find chimeras between different types of RNA. Please refer to Indexes and run align-for-chimeras with the smallRNA and targetRNA of your choice.
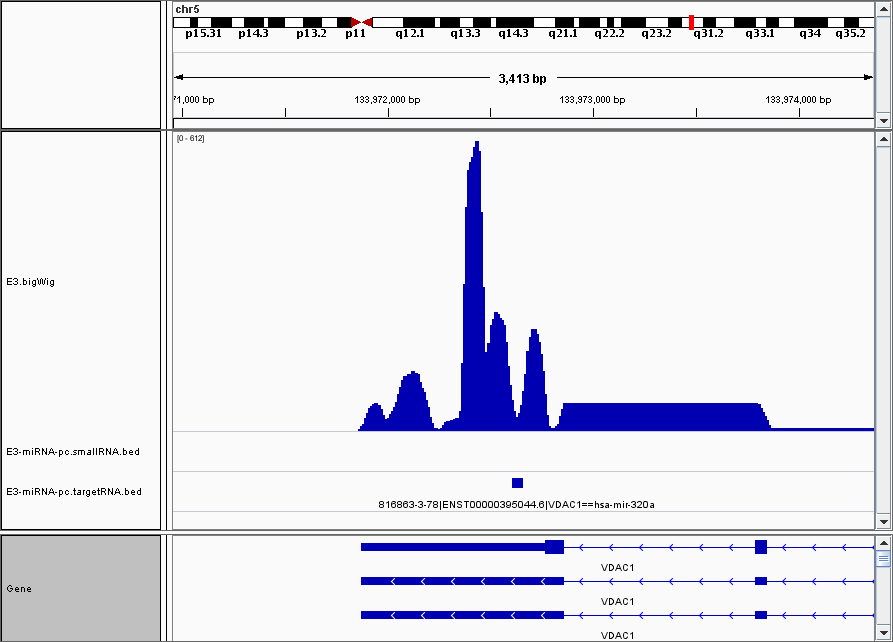
Visualisation in Genome Browser¶
This is an example visualization in IGV with the normalized coverage included as a track
Chimeras table¶
Here is the example chimeras table that is generated. The columns information can be found commented in the first lines
Issues & Feedback¶
If you encounter any issues, please report it on the Issues page of the Github repository. Please feel free to offer your suggestions and feedback and contribute by submitting pull requests.